COVID-19 mRNA "Vaccines" aka Gene Therapy Products: Lessons Learned from the Registrational Trials and Global Vaccination Campaign | Researchers Call For A Global Moratorium On mRNA Injections

The Expose | Rhoda Wilson | Expose-News.org
In a paper published on Wednesday, researchers re-analysed the Pfizer covid “vaccine” phase 3 trial data and found more serious adverse events among those in the vaccine group.
This is not what published reports from Pfizer’s phase 3 trials said. “Many key trial findings were either misreported or omitted entirely from published reports,” the researchers said.
Seven researchers – M. Nathaniel Mead, Stephanie Seneff, Russ Wolfinger, Jessica Rose, Kris Denhaerynck, Steve Kirsch and Peter A. McCullough – set out to re-analyse Pfizer’s trial data because:
- our understanding of covid vaccinations and their impact on health and mortality has evolved substantially since the first vaccine rollouts; and,
- problems with the methods, execution, and reporting of the pivotal phase 3 trials have emerged.
On Wednesday, they published their findings in a peer-reviewed papertitled ‘Covid-19 mRNA Vaccines: Lessons Learned from the Registrational Trials and Global Vaccination Campaign’. The paper was published in Cureus, a journal of medical science.
“Re-analysis of the Pfizer trial data identified statistically significant increases in serious adverse events (SAEs) in the vaccine group,” the researchers wrote.
Adding, “Numerous SAEs were identified following the Emergency Use Authorisation (EUA), including death, cancer, cardiac events, and various autoimmune, haematological, reproductive, and neurological disorders.”
The EUA the researchers are referring to is the authorisation granted to Pfizer by the US Food and Drugs Administration (“FDA”).
As the paper noted, Pfizer’s covid “vaccines” never underwent adequate safety and toxicological testing according to previously established scientific standards. It goes on to detail the absolute risk reduction, the underreporting of harms during trials, the shifting narratives and illusions of protection, quality control and manufacturing process-related impurities, the biological mechanisms underlying adverse events (“AEs”) and why, based on how our immune systems work, the vaccine is ineffective.
Concluding their comprehensive review, the researchers wrote:
Given the extensive, well-documented SAEs and unacceptably high harm-to-reward ratio, we urge governments to endorse a global moratorium on the modified mRNA products until all relevant questions pertaining to causality, residual DNA, and aberrant protein production are answered.Mead M, Seneff S, Wolfinger R, et al. (January 24, 2024) COVID-19 mRNA Vaccines: Lessons Learned from the Registrational Trials and Global Vaccination Campaign. Cureus 16(1): e52876. doi:10.7759/cureus.52876
The paper noted that the gene therapy products (“GTPs”) vaccine platform has been studied for over 30 years as an experimental cancer treatment, with the terms “gene therapy” and “mRNA vaccination” often used interchangeably.
“Although we employ the terms ‘vaccine’ and ‘vaccination’ throughout this paper, the covid-19 mRNA products are also accurately termed gene therapy products (GTPs) because, in essence, this was a case of GTP technology being applied to vaccination,” they wrote.
As such, throughout their analysis, the terms “vaccines” and “vaccinations” are used interchangeably with injections, inoculations, biologicals, or simply, products.
The following are some excerpts from the paper. You can read the full paper:
 Learn more
Learn more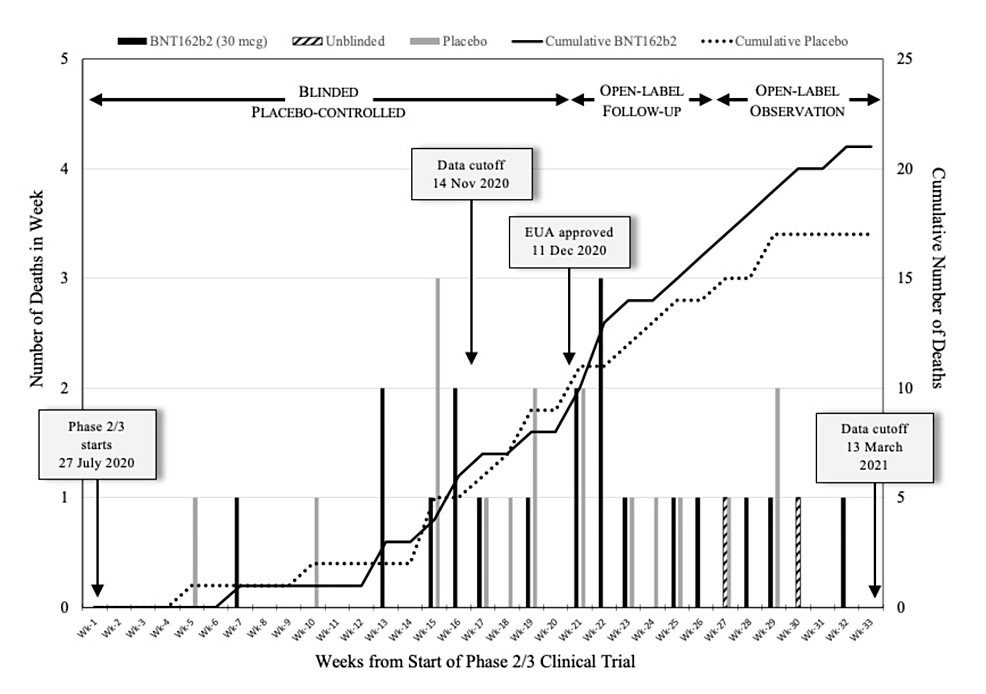
Serious Harms Revealed after EUA was Granted
In this narrative review, we revisit the registrational trials and review analyses of the AEs from these trials and other relevant studies. Most of the revelations have only recently come to light, due to the past few years of extensive censorship of healthcare professionals and research scientists who challenged the prevailing narrative set forth by the vaccine enterprise.
Despite the rhetoric, no large randomised double-blind placebo-controlled trials have ever demonstrated reductions in SARS-CoV-2 transmission, hospitalisation or death.
The study designs for the pivotal trials that led to the EUA were never intended to determine whether the mRNA inoculations could help prevent severe disease or premature death.
It was only after the EUA that the serious biological consequences of rushing the trials became evident, with numerous cardiovascular, neurological, reproductive, haematological, malignant, and autoimmune SAEs identified and published in the peer-reviewed medical literature.
Moreover, the covid mRNA vaccines produced via Process 1 and evaluated in the trials were not the same products eventually distributed worldwide; all of the covid-19 mRNA products released to the public were produced via Process 2 and have been shown to have varying degrees of DNA contamination.
The process-related impurities were absent from the covid-19 mRNA products used in the registrational trials. Virtually all doses used in those trials originated from “clinical batches” produced using what is known as Process 1. As a post-authorisation emergency supply measure for global distribution, however, a method much more suitable for mass production known as Process 2 was devised utilising bacterial plasmid DNA.
The failure of regulatory authorities to heretofore disclose process-related impurities (e.g., SV40) has further increased concerns regarding safety and quality control oversight of mRNA vaccine manufacturing processes.
Incentives Played a Key Role in Undermining Scientific Evaluation
Political and financial incentives may have played a key role in undermining the scientific evaluation process leading up to the EUA.
Before the pandemic, the US National Institutes of Health invested $116 million (35%) in mRNA vaccine technology, the Biomedical Advanced Research and Development Authority (“BARDA”) had invested $148 million (44%), while the Department of Defence (“DOD”) contributed $72 million (21%) to mRNA vaccine development.
BARDA and the DOD also collaborated closely in the co-development of Moderna’s mRNA vaccine, dedicating over $18 billion, which included guaranteed vaccine purchases. This entailed pre-purchasing hundreds of millions of mRNA vaccine doses, alongside direct financial support for the clinical trials and the expansion of Moderna’s manufacturing capabilities.
Once the pandemic began, $29.2 billion – 92% of which came from US public funds – was dedicated to the purchase of covid-19 mRNA products; another $2.2 billion (7%) was channelled into supporting clinical trials, and $108 million (less than 1%) was allocated for manufacturing and basic research.
Using US taxpayer money to purchase so many doses in advance would suggest that, before the EUA process, US federal agencies were strongly biased toward successful outcomes for the registrational trials.
Established Vaccine Testing Period Abolished
Before the rapid authorisation process, no vaccine had been permitted for market release without undergoing a testing period of at least four years. Previous timeframes for phase 3 trial testing averaged 10 years. Health departments have stated that 10-15 years is the normal timeframe for evaluating vaccine safety.
The previously established 10-15-year timeframe for clinical evaluation of vaccines was deemed necessary to ensure adequate time for monitoring the development of AEs such as cancers and autoimmune disorders.
Pfizer’s covid vaccine completed the process in seven months.
Established Safety Standards Abolished
With the covid vaccines, safety was never assessed in a manner commensurate with previously established scientific standards, as numerous safety testing and toxicology protocols typically followed by the FDA were sidestepped.
Historical accounts bear witness to instances where vaccines were prematurely introduced to the market under immense pressure, only to reveal disabling or even fatal AEs later on. Examples include the 1955 contamination of polio vaccines, instances of Guillain-Barré syndrome observed in flu vaccine recipients in 1976, and the connection between narcolepsy and a specific flu vaccine in 2009.
Against this backdrop, it is not surprising that so many medical and public health experts voiced concerns about covid mRNA vaccines bypassing the normal safety testing process.
Concerns about inadequate safety testing extend beyond the usual regulatory approval standards and practices.
As there were no specific regulations at the time of the rapid approval process, regulatory agencies quickly “adapted” the products, generalised the definition of “vaccine” to accommodate them, and then authorised them for EUA for the first time ever against a viral disease.
Due to the GTPs’ reclassification as vaccines, none of their components have been thoroughly evaluated for safety. The main concern, in a nutshell, is that the covid mRNA products may transform body cells into viral protein factories that have no off-switch – i.e., no built-in mechanism to stop or regulate such proliferation – with the spike protein (“S-protein”) being generated for prolonged periods, causing chronic, systemic inflammation and immune dysfunction.
When the S-protein enters the bloodstream and disseminates systemically, it may become a contributing factor to diverse AEs in susceptible people.
Enforce a Global Moratorium
Given the well-documented SAEs and unacceptable harm-to-reward ratio, we urge governments to endorse and enforce a global moratorium on these modified mRNA products until all relevant questions pertaining to causality, residual DNA, and aberrant protein production are answered.
Original Article: https://expose-news.com/2024/01/26/researchers-urge-a-global-moratorium-on-mrna/
Comments ()